Auswärtige Partner
PI: Thomas Ried, M. D., National Cancer Institute, National Institutes of Health, Bethesda, MD, USA
In individuellen Zellen innerhalb eines Tumors bestehen erhebliche Unterschiede im DNA-Gehalt und in der Anzahl und Struktur einzelner Chromosomen. Diese genetische Heterogenität innerhalb eines Tumors spielt nicht nur eine kritische Rolle für die Tumorprogression, sondern könnte auch dazu beitragen, dass kolorektale Karzinome so resistent gegenüber therapeutischen Interventionen sind. Intratumorale Heterogenität kann auf einer Veränderung der Kopienanzahl bestimmter Chromosomen oder einem zellindividuellen Mutationsspektrum oder beidem beruhen. Wir untersuchen deshalb in Kooperation mit der UMG-Klinik für Viszeralchirurgie (Professor Michael Ghadimi, Dr. Jochen Gaedcke), ob Heterogenität dafür verantwortlich ist, dass eine beträchtliche Anzahl von Patienten mit lokal fortgeschrittenem Rektumkarzinom nicht suffizient auf neoadjuvante Radiochemotherapie anspricht. Wir vermuten, dass unsere Analysen resistente Subklone identifizieren könnten, die unter der Therapie selektiert werden. Der Kollegiat soll Einzelzellanalysen an prätherapeutischen Biopsien von Respondern und Non-Respondern durchführen. Das geschieht durch FISH zur Identifikation von Chromosomen, deren Fehlverteilung an kolorektalen Karzinomen häufig ist. Zusätzlich führen wir Exom-Sequenzierungen an Tumorbiopsien vor und nach neoadjuvanter Radiochemotherapie durch, um das Mutationsspektrum von Genen, die für das Rektumkarzinom oder für Therapieresistenz verantwortlich sind, zu bestimmen.
Modellsysteme und Methoden, die ein Kollegiat in der Abteilung verwenden kann:
- Rho Kinase Einzelzellklone für die Analyse der Tumorheterogenität
- Fluorescent in situ hybridization (FISH)
- Zusammenarbeit mit Kollegen des NCBI zur computergestützten Modellierung
Publikationen:
Heselmeyer-Haddad K, Berroa Garcia LY, Bradley A, et al. Single-cell genetic analysis of ductal carcinoma in situ and invasive breast cancer reveals enormous tumor heterogeneity yet conserved genomic imbalances and gain of MYC during progression. The American journal of pathology 2012;181:1807-22.
Chowdhury SA, Shackney SE, Heselmeyer-Haddad K, Ried T, Schaffer AA, Schwartz R. Phylogenetic analysis of multiprobe fluorescence in situ hybridization data from tumor cell populations. Bioinformatics 2013;29:i189-i98.
Liu X, Ory V, Chapman S, et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. The American journal of pathology 2012;180:599-607.
PI: Frau Professorin Ute Moll, Stony Brook University
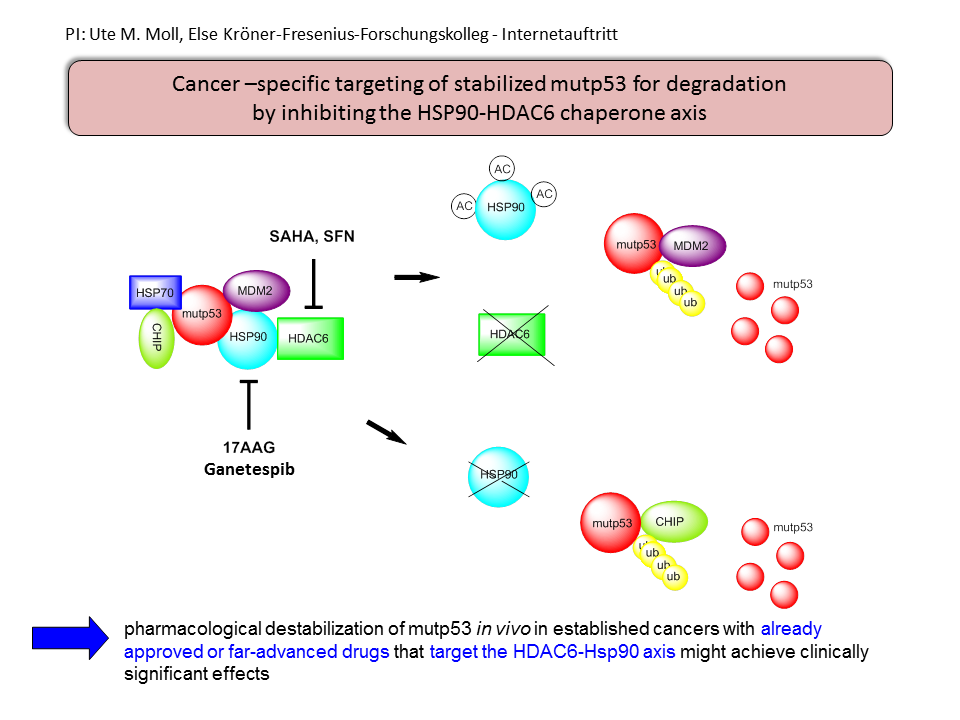
In etwa 50 % aller Tumore weist der Tumorsuppressor p53 ‚missense’ Mutationen auf, die zum einen die suppressorische Funktion von p53 inhibieren und zum anderen der neuen mutierten p53-Form (mutp53) tumorwachstumsfördernde Funktionen verleihen, den sogenannten Gain-of-function (GOF). GOF wird ermöglicht durch hohe mutp53 Proteinstabilisierung im Tumor, aber nicht in normalen Zellen. mutp53GOF verstärkt die Tumorentwicklung durch höhere Tumoraggressivität, höhere Resistenz gegen Chemotherapeutika und Metastasenbildung. Den mutp53 GOF haben wir in unserem eigenen mutp53 knock-in Mausmodell (R248Q hotspot Mutation) eindeutig gezeigt. Nun soll der mutp53 GOF in Kolonkarzinom-Mausmodellen untersucht werden. Anreicherung des mutp53 Proteins ist abhängig vom Tumorzell-aktivierten HSP90 Chaperon, welches mutp53 durch eine protektive Komplexbildung vor dem Abbau schützt. Es soll untersucht werden, ob schon etablierte Tumore eine kontinuierliche Expression von stabilisiertem mutp53 Protein für ihren Erhalt benötigen, und ob dessen Destabilisierung durch eine Inhibierung des mutp53-HSP90 Komplexes eine neue erfolgreiche Krebstherapie darstellen könnte. Die zugrunde liegenden Mechanismen des a) mutp53-HSP90 Komplexes, b) dessen Inhibierung durch HSP90 Inhibitoren, sowie der c) mutp53-vermittelten Chemoresistenz werden im Projekt identifiziert. Durch Reduktion von mutp53 mittels siRNAs soll die Abhängigkeit der Tumorzellen von mutp53 geprüft werden. Ziel ist es, mutp53 als Biomarker für die HSP90-Inhibitor-Antwort und als therapeutisches Target zu evaluieren.
Modellsysteme und Methoden, die ein Kollegiat in der Abteilung verwenden kann
-
Maus-Modelle des kolorektalen Karzinoms: transgene APC 1638N Maus, chemische AOM und/oder DSS Karzinogenese, konstitutive, teilweise humanisierte mutant p53 Knock-in Mäuse
-
Kolonoskopie an Mausmodellen, therapeutische longitudinale Langzeitstudien des Tumorverlaufes an individuellen Tieren
-
shRNA und siRNA Methoden, Chromatin Immunoprecipitation (ChIP)
Publikationen:
Li D1, Marchenko ND, Moll UM. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011 Dec;18(12):1904-13. doi: 10.1038/cdd.2011.71. Epub 2011 Jun 3.
Hanel W1, Marchenko N, Xu S, Yu SX, Weng W, Moll U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ. 2013 Jul;20(7):898-909. doi: 10.1038/cdd.2013.17. Epub 2013 Mar 29.
Dobbelstein M1, Moll U2. Targeting tumour-supportive cellular machineries in anticancer drug development. Nat Rev Drug Discov. 2014 Mar;13(3):179-96. doi: 10.1038/nrd4201.